
Protein Chemistry
We are developing powerful chemical methods to synthesize and modify proteins. In 2000, we invented the “traceless Staudinger ligation” to couple a peptide with a C-terminal phosphinothioester to one having an N-terminal azido group to form an amide bond. (An optimal reagent for mediating this ligation is diphenylphosphinomethanethiol; Sigma–Aldrich 670359.) More recently, we introduced the diazo group into chemical biology. We did so by developing a facile reaction to access diazo compounds from azides, demonstrating that the versatility of diazo compounds in 1,3-dipolar cycloadditions (“click chemistry”) greatly exceeds that of azides, showing that diazo compounds endure metabolism and enable chemoselectivity in cellulo, and discovering how to tune diazo compounds for the efficient esterification of protein carboxyl groups in water. Notably, this esterification is reversible by esterases that are endogenous in human cells. We are now using tuned diazo compounds to confer upon biologics the advantages of “prodrugs”, such as cellular permeability, enabling “gene therapy without the genes“.


Chemical Forces that Stabilize Proteins
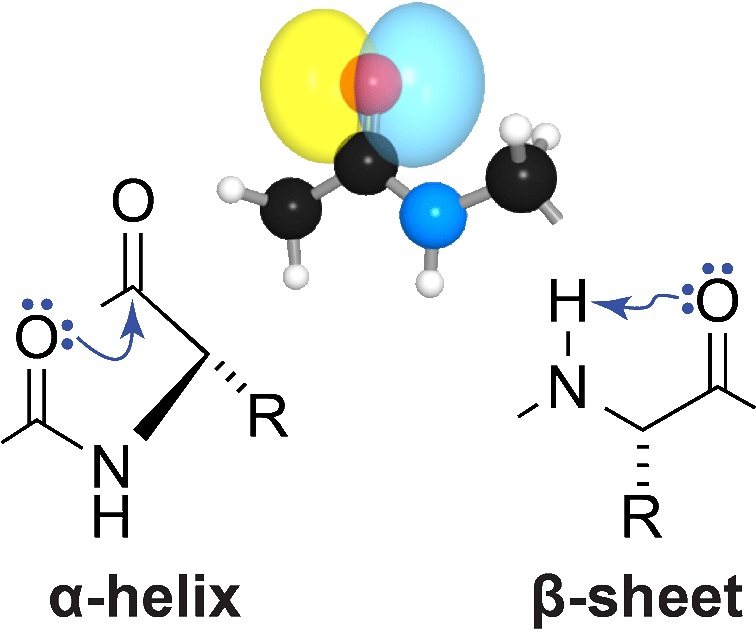
To manifest its biological activity, the amino-acid chain of a protein must fold into a particular three-dimensional structure. In the α-helices and β-sheets that dominate protein structure, a lone pair of electrons on the oxygen atom in peptide bonds accepts a hydrogen bond. Using a variety of methods from physical organic chemistry, we have shown that the other electron pair on that oxygen atom also participates in meaningful interactions in both of these architectural elements. In an α-helix, this interaction is an C=O···C=O n→π* interaction with the next carbonyl group in the main chain. In a β-sheet, this interaction is a C=O···H–N hydrogen bond within the residue. Both of these interactions entail the formation of 5-membered rings and involve significant overlap of non-bonding and anti-bonding orbitals. Whereas the canonical hydrogen bonds engage the s orbital of the oxygen, the n→π* interaction and “C5” hydrogen bond engage the p orbital, which is orthogonal to the C=O bond. These latter interactions, which are enhanced by the orbital demixing that accompanies canonical hydrogen-bond formation, have measurable effects on protein structure. We are now exploring the consequences of these unappreciated forces for the folding, stability, and aggregation of proteins.


{kind=link}
{kind=link}