
Ribonuclease: From kcat/KM to the Clinic
By understanding the underlying cellular processes, we are able to endow an otherwise innocuous human enzyme with clinically useful cytotoxicity. Ribonucleases catalyze the cleavage of RNA. Early-on, we made contributions to chemical enzymology (e.g., general acid–base catalysis and enzymatic processivity) and protein folding (e.g., prolyl peptide-bond isomerization and disulfide-bond formation) by using ribonuclease A as a model system. Then, we reasoned that this cationic enzyme, which can enter human cells (which are anionic), could become cytotoxic if it were able to evade a cytosolic inhibitor protein. By determining the structure of the human enzyme·inhibitor complex and making rational changes to a handful of amino acid residues, we have created ribonucleases that evade the inhibitor and are indeed toxic to human cells, verifying our hypothesis. We have also demonstrated that ribonucleases have a marked preference for killing cancerous cells due to a nanomolar affinity for Globo H, which is a cell-surface hexasaccharide that is a human cancer antigen.

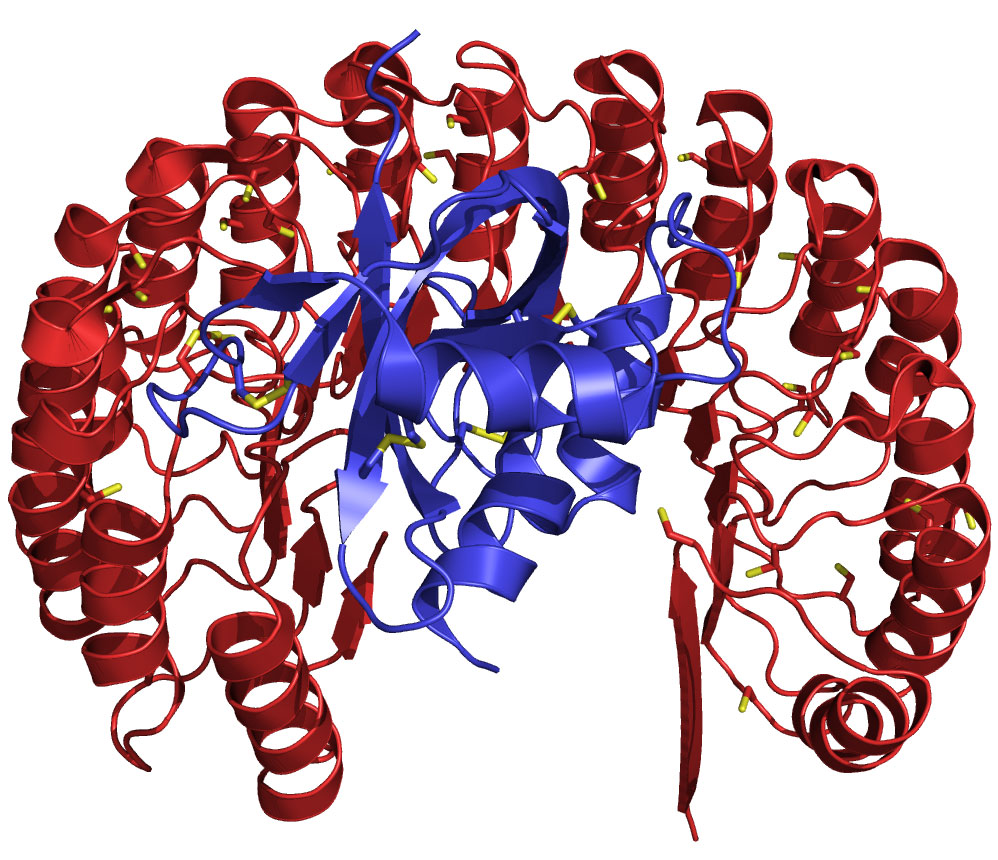
Structure of human ribonuclease (blue) bound to its natural inhibitor protein (red). This complex (Kd = 10–16 M) is the tightest known between biomolecules, and is conserved in vertebrates. The functional diversification of ribonucleases illuminates aspects of vertebrate evolution, including the regulation of extracellular RNA.
An inhibitor-evading variant of the human ribonuclease is in a Phase I clinical trial as a cancer chemotherapeutic agent. Over 50 patients have been treated with our ribonuclease at the University of Texas MD Anderson Cancer Center and the University of Wisconsin Carbone Cancer Center, and many of these patients have achieved stable disease. To enable on‑going mechanistic analyses, we have used CRISPR/Cas9 to knockout the inhibitor protein in HeLa cells, making them defenseless against invading ribonucleases. We are also determining whether natural N‑glycosylation enables the ribonuclease in human serum to evade the inhibitor protein and provide an innate immunity against cancer, and we are developing new small-molecule fluorogenic probes to illuminate the pathway taken by extracellular ribonucleases towards intracellular RNA.
Angiogenin—A Remarkable Growth Factor
How can enzyme-catalyzed RNA cleavage promote the growth of new blood vessels? The responsible enzyme—angiogenin—was the first substance shown to promote organogenesis (here, neovascularization). Its 1985 discovery at Harvard Medical School was lauded on the front page of the New York Times. Despite its importance, the mechanism by which a ribonuclease promotes cell proliferation was a mystery until we discovered that angiogenin catalyzes the cleavage of a particular phosphodiester bond in a nucleolar substrate: promoter-associated RNA (pRNA). That cleavage prevents the silencing of ribosomal DNA transcription by the nucleolar remodeling complex (NoRC). We also found that angiogenin is phosphorylated on key serine residues, enabling evasion of the ribonuclease inhibitor protein and passage to the nucleus. Thus, unlike canonical growth factors that deliver epigenetic information to DNA indirectly via receptor-mediated signal-transduction pathways, angiogenin is unique in conveying its proliferative signal from outside of the cell directly to a nucleic acid in an autonomous manner. We are now exploring the broad implications of these findings. For example, kinase inhibitors used in the clinic can keep angiogenin susceptible to its potent cytosolic inhibitor. Thus, some cancer chemotherapeutic agents could act, in part, by an unappreciated mechanism—decreasing the angiogenic activity of endogenous angiogenin.

Scheme of the cellular action of angiogenin. Angiogenin binds to syndecan-4 on the cell surface and is internalized by endocytosis. A fraction translocates to the cytosol, where angiogenin is phosphorylated by PKC and CDK. Phosphorylation endows angiogenin with the ability to evade the ribonuclease inhibitor protein. Phosphorylated angiogenin translocates into the nucleus and accumulates in the nucleolus. There, angiogenin digests pRNA, leading to the dissociation of TIP5 from the rDNA promoter. The ensuing rDNA transcription fuels the proliferation of endothelial cells (neovascularization) and tumor cells (cancer progression).
Amyotrophic lateral sclerosis (ALS or “Lou Gehrig’s Disease”) is a notorious disease for which there is no cure and but one (ineffective) drug. The disease has been linked to mutations that alter the activity of two enzymes: ↑superoxide dismutase and ↓angiogenin (which is a secretory ribonuclease). Realizing these links, we have generated a “masked” angiogenin that is activated by hydrogen peroxide, which is a product of catalysis by superoxide dismutase. We found that the masked angiogenin, unlike the wild-type enzyme, does not promote the proliferation of human endothelial cells, which would be undesirable. The masked enzyme does, however, protect human astroglia from oxidative stress, which is highly desirable. These findings validate an unprecedented attack at the core of ALS.
Gene Therapy Without the Genes
The intracellular activity of many enzymes and proteins can be decreased by small-molecule inhibitors or antagonists. Increasing intracellular activity is, however, a much more formidable challenge. In 2007, we introduced the method of “super-charging”—converting anionic residues into cationic ones so as to enhance uptake into cells (which are anionic). That method is efficient for directing proteins into endosomes but not the cytosol, is often deleterious to protein structure/function, and is not bioreversible, which could lead to immunogenicity.
Now, we are using esterification with diazo compounds to “cloak” anionic carboxylates and thereby to enable protein uptake directly into the cytosol. There, esterases convert the nascent esters back into carboxylates. This novel approach is enabling us to replace dysfunctional proteins, interfere with protein–protein interactions, control stem-cell fate, and edit the genome.

Cellular Redox Homeostasis
Making correct disulfide linkages between cysteine residues (and undoing incorrect ones) is a critical but often complex aspect of protein folding. We addressed this problem from several directions using diverse tools. Protein disulfide isomerase (PDI) and thioredoxin (Trx) are enzymes that catalyze disulfide-bond formation with a common active-site motif: Cys–Xaa–Xaa–Cys, where Xaa can vary. We are combining in vitro mechanistic studies of these two enzymes with molecular genetics to define the optimal attributes of a folding catalyst, which appear to be a disulfide bond of high reduction potential (E°′) within the active site and a solvent-exposed cysteine of low pKa. Inspired by PDI and Trx, we are creating small-molecule dithiols with low thiol pKa and high disulfide reduction potential that are efficacious organocatalysts of oxidative protein folding, both in vitro and in cellulo. Finally, we designed and synthesized dithiobutylamine (DTBA; Sigma–Aldrich 774405), which is superior to the venerable dithiothreitol (DTT) as a biochemical reducing agent. We are now using DTBA conjugates to deliver electrons into physiological systems with surgical precision.

{kind=link}
{kind=link}
{kind=link}